

![]()
![]()
The vcfppR package implements various powerful functions for fast genomics analyses with VCF/BCF files using the C++ API of vcfpp.h.
## install.package("vcfppR") ## from CRAN
remotes::install_github("Zilong-Li/vcfppR") ## from latest githubIf you find it useful, please cite the paper
library(vcfppR)
citation("vcfppR")All functions in vcfppR support URL as filename of VCF/BCF files.
phasedvcf <- "https://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000G_2504_high_coverage/working/20220422_3202_phased_SNV_INDEL_SV/1kGP_high_coverage_Illumina.chr21.filtered.SNV_INDEL_SV_phased_panel.vcf.gz"
rawvcf <- "https://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000G_2504_high_coverage/working/20201028_3202_raw_GT_with_annot/20201028_CCDG_14151_B01_GRM_WGS_2020-08-05_chr21.recalibrated_variants.vcf.gz"
svfile <- "https://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000G_2504_high_coverage/working/20210124.SV_Illumina_Integration/1KGP_3202.gatksv_svtools_novelins.freeze_V3.wAF.vcf.gz"
popfile <- "https://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000G_2504_high_coverage/20130606_g1k_3202_samples_ped_population.txt"vcftable: read
VCF as tabular datavcftable gives you fine control over what you want to
extract from VCF/BCF files.
Read SNP variants with GT format and two samples
library(vcfppR)
res <- vcftable(phasedvcf, "chr21:1-5100000", samples = "HG00673,NA10840", vartype = "snps")
str(res)
#> List of 10
#> $ samples: chr [1:2] "HG00673" "NA10840"
#> $ chr : chr [1:194] "chr21" "chr21" "chr21" "chr21" ...
#> $ pos : int [1:194] 5030578 5030588 5030596 5030673 5030957 5030960 5031004 5031031 5031194 5031224 ...
#> $ id : chr [1:194] "21:5030578:C:T" "21:5030588:T:C" "21:5030596:A:G" "21:5030673:G:A" ...
#> $ ref : chr [1:194] "C" "T" "A" "G" ...
#> $ alt : chr [1:194] "T" "C" "G" "A" ...
#> $ qual : num [1:194] 2.14e+09 2.14e+09 2.14e+09 2.14e+09 2.14e+09 ...
#> $ filter : chr [1:194] "." "." "." "." ...
#> $ info : chr [1:194] "AC=74;AF=0.0115553;CM=0;AN=6404;AN_EAS=1170;AN_AMR=980;AN_EUR=1266;AN_AFR=1786;AN_SAS=1202;AN_EUR_unrel=1006;AN"| __truncated__ "AC=53;AF=0.00827608;CM=1.78789e-05;AN=6404;AN_EAS=1170;AN_AMR=980;AN_EUR=1266;AN_AFR=1786;AN_SAS=1202;AN_EUR_un"| __truncated__ "AC=2;AF=0.000312305;CM=3.21821e-05;AN=6404;AN_EAS=1170;AN_AMR=980;AN_EUR=1266;AN_AFR=1786;AN_SAS=1202;AN_EUR_un"| __truncated__ "AC=2;AF=0.000312305;CM=0.00016985;AN=6404;AN_EAS=1170;AN_AMR=980;AN_EUR=1266;AN_AFR=1786;AN_SAS=1202;AN_EUR_unr"| __truncated__ ...
#> $ gt : int [1:194, 1:2] 0 0 0 0 0 0 0 0 0 0 ...
#> - attr(*, "class")= chr "vcftable"Read all variants followed by subsetting operations
vcffile <- system.file("extdata", "raw.gt.vcf.gz", package="vcfppR")
res <- vcftable(vcffile, "chr21:1-5050000")
str(subset(res, pos >= 5000000 & pos <= 5030300 & nchar(ref) == 1 & nchar(alt) == 1))
#> List of 10
#> $ samples: chr [1:3202] "HG00096" "HG00097" "HG00099" "HG00100" ...
#> $ chr : chr [1:5] "chr21" "chr21" "chr21" "chr21" ...
#> $ pos : int [1:5] 5030082 5030088 5030105 5030253 5030278
#> $ id : chr [1:5] "chr21:5030082:G:A" "chr21:5030088:C:T" "chr21:5030105:C:A" "chr21:5030253:G:T" ...
#> $ ref : chr [1:5] "G" "C" "C" "G" ...
#> $ alt : chr [1:5] "A" "T" "A" "T" ...
#> $ qual : num [1:5] 70.1 2773.1 3897.8 102.6 868.9
#> $ filter : chr [1:5] "VQSRTrancheSNP99.80to100.00" "VQSRTrancheSNP99.80to100.00" "VQSRTrancheSNP99.80to100.00" "VQSRTrancheSNP99.80to100.00" ...
#> $ info : chr [1:5] "AC=2;AF=0.000616523;AN=3244;DP=2498;FS=0;MLEAC=1;MLEAF=0.0003083;MQ=17.07;MQ0=0;QD=17.52;SOR=3.258;VQSLOD=-32.6"| __truncated__ "AC=127;AF=0.0400126;AN=3174;BaseQRankSum=0.736;ClippingRankSum=0.736;DP=2750;FS=0;InbreedingCoeff=0.0015;MLEAC="| __truncated__ "AC=128;AF=0.0352811;AN=3628;BaseQRankSum=0.736;ClippingRankSum=0.727;DP=3476;FS=0;InbreedingCoeff=-0.0015;MLEAC"| __truncated__ "AC=1;AF=0.000165837;AN=6030;BaseQRankSum=-0.583;ClippingRankSum=-0.259;DP=19530;FS=0;InbreedingCoeff=-0.0274;ML"| __truncated__ ...
#> $ gt : int [1:5, 1:3202] NA 2 2 0 0 0 NA NA 0 0 ...
#> - attr(*, "class")= chr "vcftable"Read INDEL variants with DP format and QUAL>100
res <- vcftable(rawvcf, "chr21:1-5100000", vartype = "indels", format = "DP", qual=100)
vcfplot(res, which.sample = 10, ylim=c(0,60), col = 3, pch=19)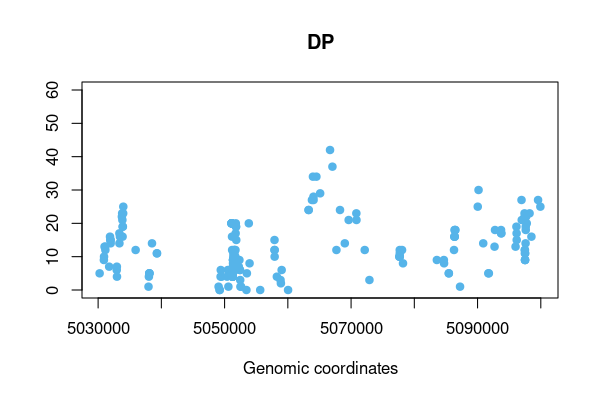
vcfcomp:
compare two VCF files and report concordanceWant to investigate the concordance between two VCF files?
vcfcomp is the utility function you need! For example, in
benchmarkings, we intend to calculate the genotype correlation between
the test and the truth.
res <- vcfcomp(test = rawvcf, truth = phasedvcf,
stats = "r2", region = "chr21:1-5100000",
formats = c("GT","GT"), setid = TRUE)
par(mar=c(5,5,2,2), cex.lab = 2)
vcfplot(res, col = 2,cex = 2, lwd = 3, type = "b")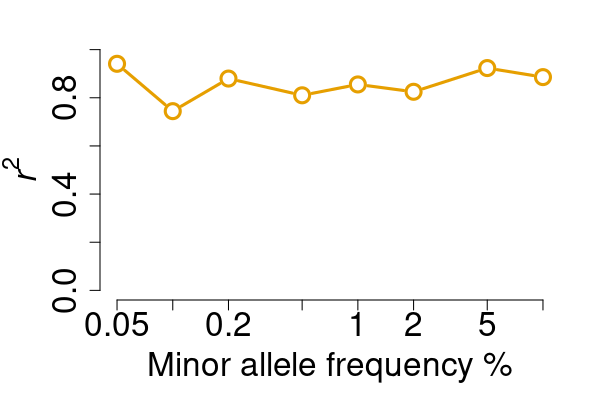
Also, we want to investigate the relationship between the genotype concordance and the call rate ranked by the genotype quality.
svvcf <- system.file("extdata", "platinum.sv.vcf.gz", package="vcfppR")
svuppvcf <- system.file("extdata", "svupp.call.vcf.gz", package="vcfppR")
truth <- vcftable(svvcf)
truth$neighbors <-as.integer(sub(".*NumNeighbors=([^;]+).*", "\\1", truth$info))
truth <- subset(truth, neighbors == 0) ## subset biallelic SVs
res <- vcfcomp(svuppvcf, truth, stats = "gtgq", region = "chr1")
vcfplot(res, col = 2,cex = 2, lwd = 3, type = "l", bty = 'l')
Check out the vignettes for more!
vcfsummary:
variants characterizationWant to summarize variants discovered by genotype caller e.g. GATK?
vcfsummary is the utility function you need!
Small variants
res <- vcfsummary(rawvcf,"chr21:10000000-10010000")
vcfplot(res, pop = popfile, col = 1:5, main = "Number of SNP & INDEL variants")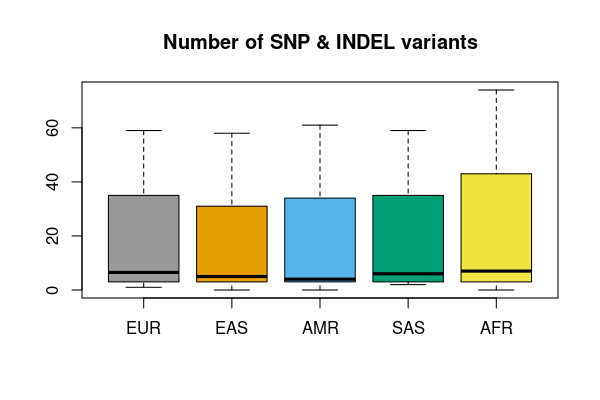
Complex structure variants
res <- vcfsummary(svfile, svtype = TRUE, region = "chr20")
vcfplot(res, main = "Structure Variant Counts", col = 1:7)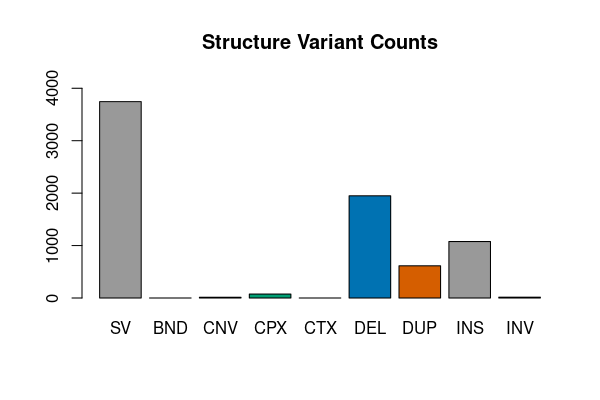
There are two classes i.e. vcfreader and
vcfwriter offering the full R-bindings of
vcfpp.h. Check out the examples in the tests folder or refer to the manual,
e.g. ?vcfppR::vcfreader.
library(testthat)
svfile <- system.file("extdata", "sv.vcf.gz", package="vcfppR")
test_that("can change samples name and set genotypes for single sample", {
br <- vcfreader$new(svfile, "", "HG00096")
br$variant()
expect_identical(br$infoStr("SVTYPE"), "DUP")
expect_identical(br$genotypes(F), c(0L, 0L))
br$setGenotypes(c(1L,1L))
expect_identical(br$genotypes(F), c(1L, 1L))
outfile <- paste0(tempfile(), ".vcf.gz")
br$output(outfile)
br$updateSamples("ZZZZZ")
br$write()
br$close()
vcf <- vcftable(outfile)
expect_true(vcf$gt==2)
expect_true(vcf$samples=="ZZZZZ")
})
#> Test passed 🎉